Thalassemia
Thalassemia major occurs when a child inherits two mutated genes, one from each parent. Children born with thalassemia major usually develop the symptoms of severe anemia within the first year of life. They lack the ability to produce normal, adult hemoglobin and experience chronic fatigue. They may also fail to thrive.
Two major consequences of the genetic defect of thalassemia are severe anemia and expansion of the bone marrow in the body�s effort to produce more red blood cells. This leads to poor growth, impaired physical activities, facial and other bone deformities, fragile bones and enlargement of the liver and spleen. If left untreated, it will lead to death within the first decade of life. The only treatment to combat severe anemia is regular blood transfusions and iron chelation therapy.
What is it?
Thalassemia is a group of genetic blood diseases that vary widely in severity involving decreased and defective production ofhemoglobin, a molecule that's found inside all red blood cells and is necessary to transport oxygen throughout the body.
Hemoglobin contains two different kinds of protein chains named alpha and beta chains. Any deficiency in these chains causes abnormalities in the formation, size, and shape of red blood cells. There are two types of thalassemia: alpha-thalassemia and beta-thalassemia. Their names describe which protein chain, alpha or beta, in the hemoglobin molecule is abnormal. Both forms of thalassemia are genetic disorders and both parents have to pass the thalassemia gene to their child for him or her to have the disease.
If only one parent passes the gene for thalassemia on to their child, then the child is said to have thalassemia trait. Thalassemia trait will not develop into the full-blown disease, has no or few symptoms and no treatment is necessary for someone who has it. However, genetic counseling is important for families that carry the thalassemia gene because someone with the trait has a 25% (1 in 4) chance of having a child with the disease if his or her partner also carries the trait. Thalassemia affects many people coming from areas around the Mediterranean (Italy, Greece, and Turkey), Africa, Malaysia, China, and many parts of Southeast Asia.
Thalassemia can cause ineffective production of red blood cells in addition to hemolysis (destruction of red blood cells). People with different forms of thalassemia show a wide range of illness from the disease. Some people only have mild anemia with little or no effects, whereas others require frequent blood transfusions.
How is it diagnosed?
A complete blood count, hemoglobin and a specific blood test called a hemoglobin electrophoresis is used to screen for all types of thalassemia and can be done in infancy.
What does it look like?
Alpha-Thalassemia
Alpha-thalassemia can range from mild to severe. Children with alpha-thalassemia trait do not have thalassemia disease. They have no significant health problems, with the exception of possibly being mildly anemic. The anemia may cause slight fatigue, and doctors may at first suspect an iron deficiency. Other cases of alpha-thalassemia resemble beta-thalassemia intermedia (see next section). Finally, the most severe form of the disorder is called alpha-thalassemia major. This type is extremely rare and is sometimes incompatible with life. Women carrying fetuses with this form of thalassemia have a high incidence of miscarriage because the fetuses cannot survive.
How is it treated?
Iron doesn't help children with thalassemia trait, however, because the anemia is caused by lowered hemoglobin levels, not a lack of iron. People with alpha-thalassemia intermedia may require occasional blood transfusions during times of physical stress, like fevers or other illnesses.
Beta-Thalassemia
Beta-thalassemia, the most common form of the disorder seen in the Canada and the United States, is grouped into three categories: beta-thalassemia minor (trait), intermedia, and major (Cooley's anemia).
Beta-thalassemia minor (trait)
Beta-thalassemia minor often goes undiagnosed because people with the condition have no real symptoms other than mild anemia. No treatment is usually needed.
Beta-thalassemia intermedia
Children with beta-thalassemia intermedia have varying effects from the disease. They tend to have similar findings but a milder course and better outlook than do those with thalassemia major. Mild anemia might be their only symptom or they might require blood transfusions, especially during illness, medical complications, or later on during pregnancy.
Beta-thalassemia major (Cooley's anemia) This is a severe condition in which regular blood transfusions are necessary for the child to survive. Without transfusions every few weeks, children diagnosed with thalassemia major would usually die by age 7 due to the effects of severe anemia on the body.

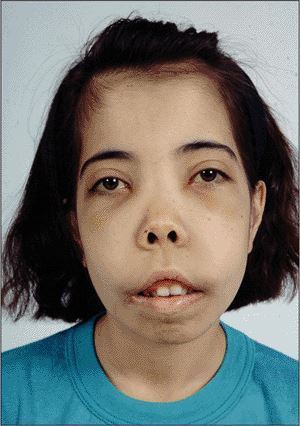
What does it look like?
The most common complaint is fatigue or shortness of breath. Some children also have mild jaundice or the liver and the spleen may be enlarged, which is sometimes painful.
Another symptom of beta-thalassemia major can be bone abnormalities. Because the bone marrow is working overtime to make more red blood cells to counteract the anemia, children can experience enlargement of their cheek bones, foreheads, and other bones. They may experience bone pain or frequent bleeding nose. Gallstones are also a frequent complication of thalassemia because of abnormalities in bile production that involve the liver and the gallbladder. Poor growth may occur as a result of low hemoglobin and reduced ability of the blood to carry oxygen to the body. Regular transfusions can help alleviate these problems. Heart failure and infection are the leading causes of death among children with untreated thalassemia major.
How is it treated?
Although multiple lifelong transfusions save lives, they also cause a serious side effect: an overload of iron in the bodies of people with thalassemia. Over time, people with thalassemia accumulate deposits of iron in their bodies, especially in the liver, heart, and endocrine (hormone-producing) glands. The deposits eventually cause abnormal heart rhythms or heart failure. In addition, permanent liver scarring, diabetes, and delayed growth and sexual maturation can also occur. To minimize iron deposits in the body, people affected by the disease must undergo chelation (iron-removing) therapy for up to 12 hours a day with intravenous (given through the skin directly into a vein) or subcutaneous (under the skin) doses of the iron-binding agent desferrioxamine. Before treatment of iron-chelation therapy was widely used, most children with beta-thalassemia major died before reaching adulthood. Chelation therapy is given 5 to 7 days a week and has been proven to prevent liver and heart damage from iron overload, allow for normal growth and sexual development in children with thalassemia, and increase life span. Iron concentrations in the body are monitored every few months and are used to guide the frequency and dosing of chelation therapy. Removal of the spleen may also be recommended because the condition can cause that organ to become extremely enlarged. Children with thalassemia also have an increased need for daily folic acid. Recently, some children have successfully undergone bone marrow transplants to treat thalassemia major; however, this is considered only in cases of severely disabling thalassemia disease.
Thrombocytopathy is an abnormality of platelets. It may be congenital or acquired. It may cause a thrombotic or a bleeding tendency or may be part of a wider disorder such as myelodysplasia. Under normal circumstances, when the endothelial cells lining blood vessels are breached, platelets interact with von Willebrand factor (vWF) via the membrane glycoprotein 1b complex to help seal the breach. Glycoprotein IIb/Ia complex attracts other platelets, which combine to form aggregates.[1] The platelets contain granules which break down to release fibrinogen, vWF, platelet-derived growth factor adenosine 5'-diphosphate (ADP), calcium and 5-hydroxytryptamine (5-HT) - serotonin. All this helps to promote the formation of a haemostatic plug (primary haemostasis).
Activated platelets also synthesise thromboxane A2 from arachidonic acid as well as presenting negatively charged phospholipids on the outer leaflet of the platelet membrane bilayer. This negative surface provides binding sites for enzymes and cofactors of the coagulation system. The total effect is therefore to stimulate the coagulation system to form a clot (secondary haemostasis).
Defects of the platelet system thus manifest themselves as primary haemostatic phenomena - eg, increased bleeding times, petechiae, purpura, rather than secondary haemostatic phenomena - eg, haemarthrosis, muscle haematomas. |

